本文所有计算均采用基于DFT平面波超软赝势方法,运用MaterialsStudio计算软件中的CASTEP[22](Cambridgesequentialtotalenergypack-age)模块进行计算与分析.原子的价电子组成选取为Bi6s26p3,O2s22p4,Cl3s23p5.计算时采用广义梯度近似(generalizedgradientapproximation,GGA)中的PBE(Perdew-Burke-Ernzerhof)泛函[23].计算过程中,选取平面波截断能Ecut=340eV,用Monkorst-Pack形式[24]的高对称特殊K点方法对布里渊区进行积分,K点网格对于体相计算采用6×6×3,表面模型计算采用3×3×1.在自洽场运算中,应用Pulay密度混合法[25],设置自洽精度为每原子2×10−6eV.同时几何优化时采用BFGS(Broyden-Fletcher-Goldfarb-Shanno)算法[26],总能量收敛为每原子1×10−5eV,原子平均受力小于0.3eV/nm,最大位移为2×10−4nm,最大应力为0.05GPa.
计算模型
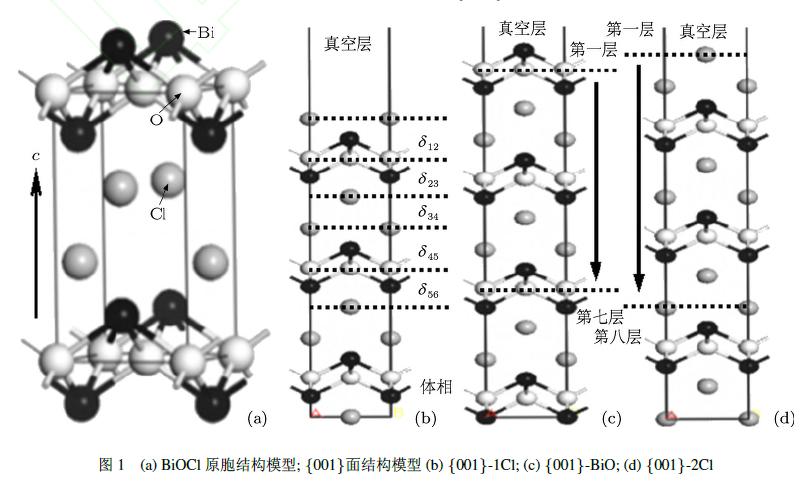
BiOCl晶体属四方晶系,空间群为P4/nmm,晶格常数为[27]a=b=3:890A°,c=7:370A°,a=b=g=90◦.BiOCl晶体结构沿c轴方向,由[Bi2O2]层与Cl原子层交错排列形成的层状正方氟氯铅矿(PbFCl)结构,其晶体结构模型如图1(a)所示.在原胞的基础上根据表面原子和终止方式的不同,理想的BiOCl{001}表面存在三种不同形式的表面构型.为了方便区分,在此定义模型I:表面终止原子为单层Cl,即为{001}-1Cl表面,共有9个原子层;II:表面终止原子为BiO,即为{001}-BiO表面,共有10个原子层;III:表面终止原子为双层Cl,即为{001}-2Cl表面,共有11个原子层.为了消除上下两个表面的相互作用,建立三种{001}表面的真空层,均沿c轴方向且厚度为15A°,{001}-1Cl,{001}-BiO,{001}-2Cl表面的结构模型分别见图1(b)—(d).计算过程中将表面模型底部的三层原子固定,让其保持BiOCl体相结构,允许模型中其余原子层自由弛豫.
|